
En breve publicaremos una nueva entrada
El sistema inmunitario y la contaminación ambiental son los inductor de los cánceres de pulmón en personas no fumadoras
En esta visión de Panorama os presento una idea revolucionaria sobre la formación de tumores en la que está implicada la respuesta inmunitaria. Y me baso en un estudio que posiblemente cambiará la forma en que vemos el cáncer de pulmón en personas que nunca han fumado. IntroducciónEl cáncer es la causa principal de mortalidad en el mundo desarrollado. Muchos…
Nuevas implicaciones de los eritrocitos en la respuesta inmunitaria y su influencia en la patogénesis de la covid-19
Introducción Clásicamente, los eritrocitos al ser células carentes de orgánulos y de información genética, se han considerado como meras bolsas lipídicas conteniendo hemoglobina e implicadas solamente en el transporte de gases por la sangre. Pero, para los inmunólogos son mucho más que células imprescindibles para la respiración, pues existe un gran vínculo entre los componentes del sistema inmunitario y los…
Mi primera lección de inmunología. El papel fisiológico del sistema inmunitario
Os presento en estas líneas lo que considero debería ser la primera lección de cualquier curso de inmunología, abordar el papel fisiológico del sistema inmunitario. Cuando en fisiología explicamos cualquier sistema de órganos, siempre empezamos haciendo una enumeración tanto de componentes como de funciones. Y allí empezamos a descubrir que las funciones que desempeñan son mucho más extensas de lo…
Las nuevas variantes del SARS-CoV-2 que nadie busca y que están haciendo al virus más resistente a la respuesta inmunitaria
Introducción Desde la aparición del SARS-CoV-2 y su rápida propagación por todo el mundo, han surgido nuevas variantes con interés médico, inmunológico y epidemiológico pues estas podrían ser más patogénicas, evadir con más éxito la respuesta inmunitaria o hacer que las vacunas fueran menos efectivas. Habida cuenta que la proteína Spike (S) del coronavirus es crucial para la infección efectiva…

El sistema inmunitario está detrás de los tatuajes
La ciencia detrás de los tatuajesA mucha gente le gusta decorar su cuerpo con tatuajes permanentes. Los tatuajes están de moda, y los que los llevan los suelen lucir con gusto y orgullo, pues muchos de ellos son auténticas piezas de arte o mensajes muy relevantes para los portadores. Los tatuajes existen gracias a que tenemos artistas y a que…
Los retos futuros de las vacunas contra la covid-19 y SARS-CoV-2
Llevamos un año de pandemia y el mundo ha hecho un esfuerzo tremendo, con mayor o menor acierto, para minimizar las devastadoras consecuencias de la covid-19. En este sentido, el mayor avance que ha generado la ciencia y la tecnología es el desarrollo meteórico de vacunas que disminuyen la gravedad de la infección y, esperemos, la transmisión de la enfermedad.…
Reflexiones sobre la pandemia covid-19 tras un año desde su brote en occidente
Este inmunoensayo estará disponible el 17 de marzo tras la clase inaugural que impartiré en la Escuela de Salud del Ateneo de Valencia
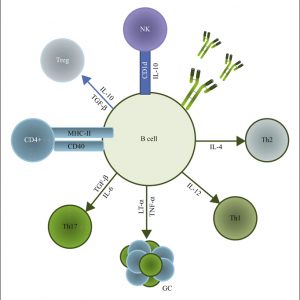
El sistema inmunitario es un supersistema
En el post anterior vimos que la complejidad del mundo microbiano repercute directamente sobre la complejidad estructural y funcional del sistema que se encarga de combatirlo. Pero la complejidad del sistema inmunitario no solo se debe a la variedad del mundo microbiano sino a la naturaleza misma que rige la generación de la inmunidad.El sistema inmunitario no opera de forma…
La inmunología, un reto imprescindible para una medicina moderna
La inmunología es una disciplina fundamental para los estudiantes de medicina, ciencias de la vida y biotecnología, que tienen que estudiar como el sistema inmunitario se organiza y funciona para protegernos y defendernos de la enfermedad. Además de su relación con la patología humana son muy relevantes sus aplicaciones diagnósticas, terapéuticas y tecnológicas. Cuando abordamos el estudio de la inmunología…